
Simulating molecular dynamics without having to compromise
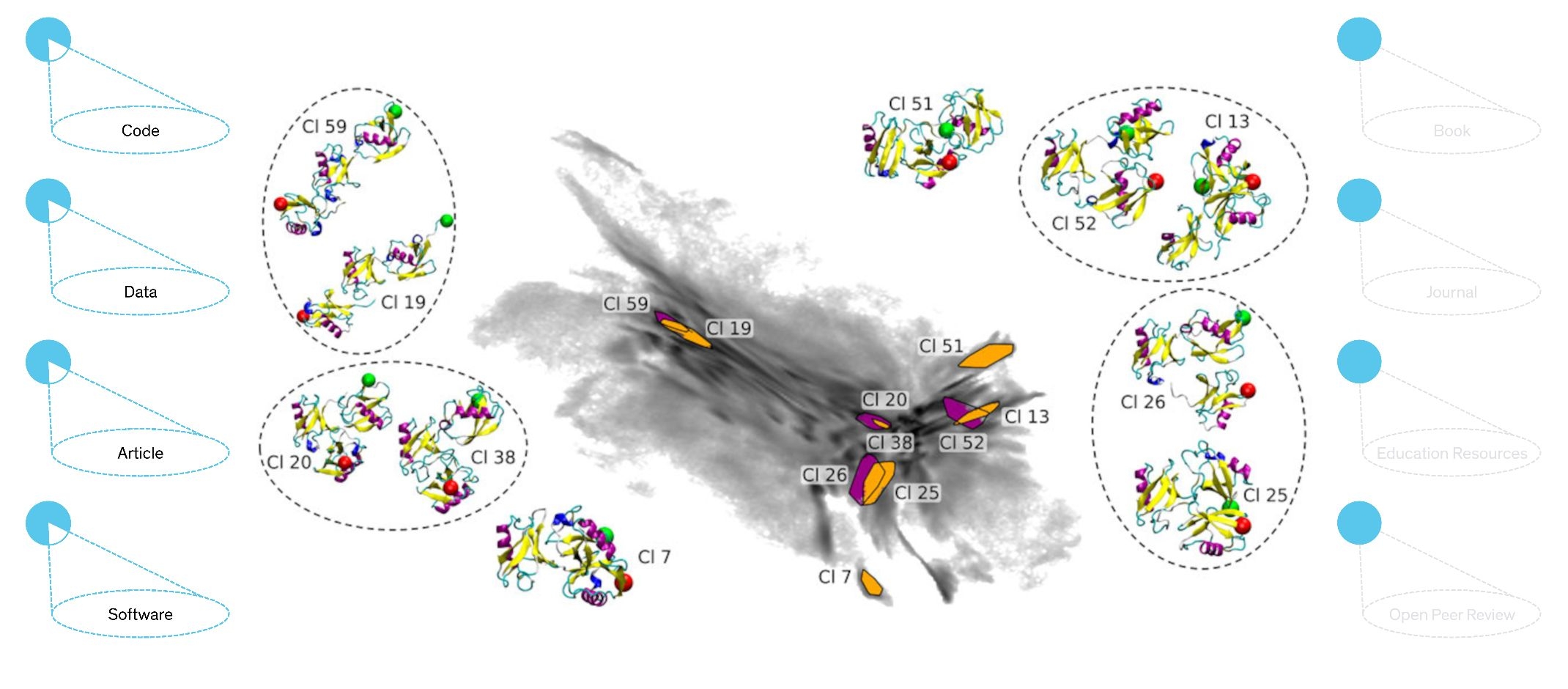
To gain deeper knowledge of the function of proteins, it is often necessary to understand how they change their spatial structure when interacting with other molecules. For this purpose, researchers use very computationally-intensive simulations. In order to simulate changes in protein structure over biologically relevant periods of time, researchers often have to make compromises: Instead of using a resolution at the atomic level, they combine several atoms into tiny "beads" to allow for longer durations of the simulated time periods.
Konstanz-based chemists Simon Hunkler, Teresa Buhl, Oleksandra Kukharenko and Christine Peter have developed a method that makes up for this loss of information. For their Back-Mapping Based Sampling (BMBS), the coarse-grained simulations are used as starting points for additional, shorter simulations that sample information at the atomic level in a more targeted way. For the most part, this method preserves the advantage of saving computer time. The researchers have shown in a recent open access article, that their method can also be applied to complex protein systems. They have freely published their results along with the analysis scripts and sample data they used.
The open access article in Frontiers in Chemistry (doi: 10.3389/fchem.2022.1087963) is available for free download from the University of Konstanz document server KOPS.
The full replication dataset for the article (doi: 10.48606/40) can be downloaded for free.
The Python notebooks used for the BMBS workflows and a minimal example are available for free download from the authors' GitHub directory.
A central component of the BMBS workflow is dimension reduction using the EncoderMap software. The software was developed in Christine Peter's research team and is available for free download from GitHub.