
Moleküldynamik simulieren ohne Kompromisse
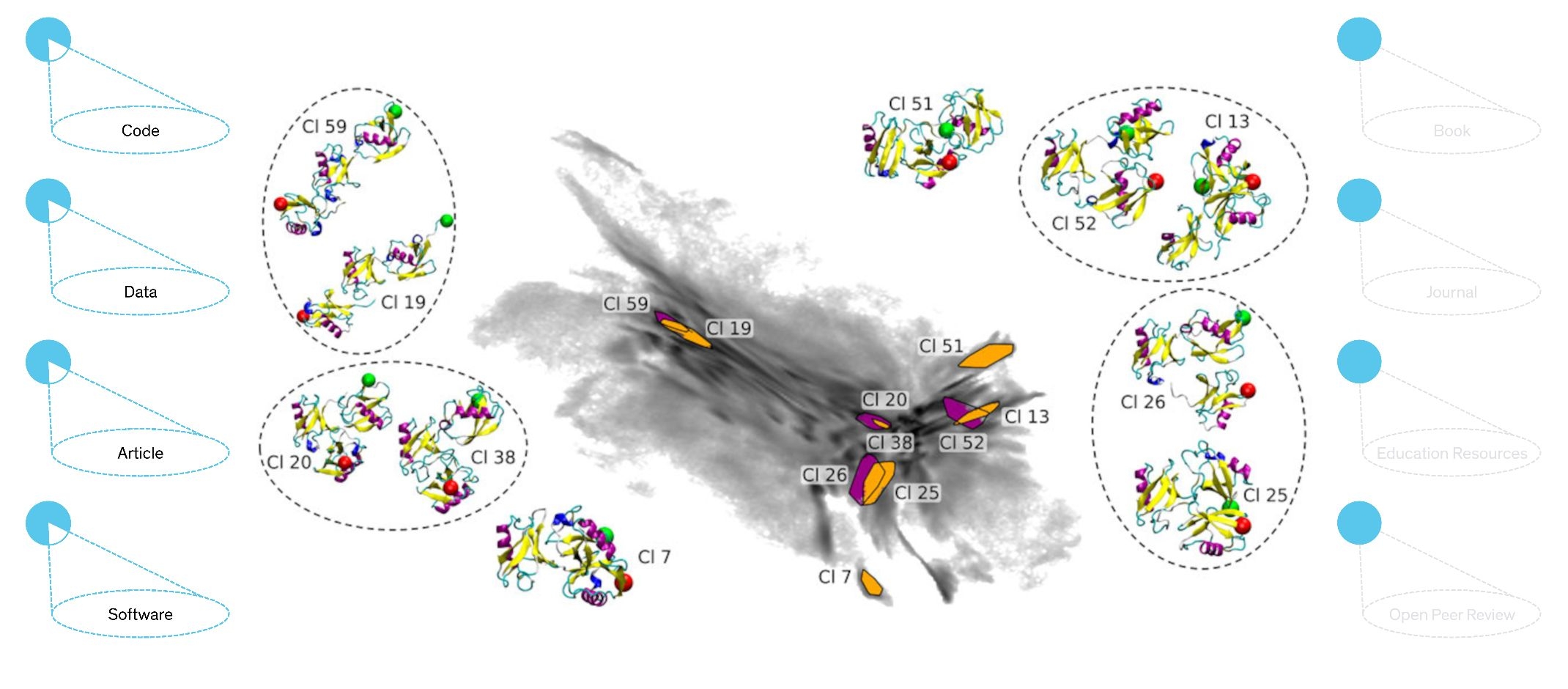
Für ein tieferes Verständnis der Funktion von Proteinen ist es oft notwendig, nachzuvollziehen, wie diese ihre räumliche Struktur verändern, wenn sie mit anderen Molekülen wechselwirken. Hierfür verwendet die Forschung sehr rechenaufwändige Computersimulationen. Um die räumlichen Veränderungen der Proteinstruktur über biologisch relevante Zeiträume simulieren zu können, wird dabei häufig ein Kompromiss eingegangen: Zugunsten der Dauer des simulierten Zeitraums wird durch Zusammenfassen von jeweils mehreren Atomen zu kleinen „Körnchen“ (engl.: „beads“) auf eine atomare räumliche Auflösung verzichtet.
Die Konstanzer ChemikerInnen Simon Hunkler, Teresa Buhl, Oleksandra Kukharenko und Christine Peter entwickelten ein Verfahren, das diesen Informationsverlust wieder wettmacht. Dabei dient die „grobkörnige“ Simulation als Ausgangspunkt für weitere, kürzere Simulationen, welche die atomare Information gezielter abfragen – das BMBS. Der Vorteil in Bezug auf die benötigte Rechenleistung bleibt so weitestgehend erhalten. Dass die Methode auf komplexe Proteinsysteme anwendbar ist, zeigen die Forschenden in einem aktuellen Open-Access-Artikel. Sie veröffentlichen ihre Ergebnisse zusammen mit den verwendeten Analyseskripten und freien Beispieldaten.
Der Open-Access-Artikel in Frontiers in Chemistry (doi: 10.3389/fchem.2022.1087963) steht auf dem Konstanzer Online-Publikationssystem (KOPS) zum kostenlosen Download bereit.
Den vollständigen Replikations-Datensatz zum Artikel (doi: 10.48606/40) finden Sie auf KonDATA zum Download.
Die Python-Notebooks, die zum Durchführen des BMBS Workflows benötigt werden, sowie ein Minimalbeispiel können kostenlos aus dem GitHub-Verzeichnis der Autoren heruntergeladen werden.
Zentraler Bestandteil des BMBS Workflows ist eine Dimensionsreduktion mithilfe der Software EncoderMap. Diese wurde ebenfalls in der Arbeitsgruppe von Christine Peter entwickelt und ist kostenlos auf GitHub zu finden.